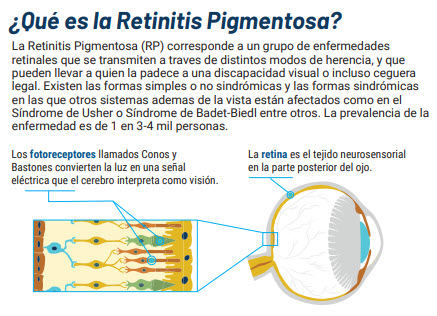
¿Qué es la Retinitis Pigmentosa?
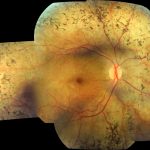
La retinitis pigmentosa es una degeneración primaria y progresiva de la retina que compromete a los fotorreceptores, siendo los bastones afectados en un comienzo seguido posteriormente por compromiso de los conos. Corresponde a una enfermedad hereditaria muy heterogénea caracterizada por ceguera nocturna de inicio en la adolescencia seguida de pérdida progresiva del campo visual periférico descrito por los pacientes como “visión en túnel”. Los hallazgos al examen clínico dependen del grado de avance de la degeneración retinal. Los cambios iniciales al examen de la retina son estrechamiento arteriolar y despigmentación del Epitelio Pigmentario Retinal (RPE). A medida que progresa el deterioro de los fotorreceptores, se evidencian acúmulos de pigmento intra-retinal en una configuración tipo “espícula ósea”. Otros signos son palidez del nervio óptico, catarata subcapsular posterior y vítreo con aspecto tipo “partículas de polvo”.
La RP se clasifica como no sindrómica o simple (sin compromiso de otros órganos o tejidos); sindrómica (en que se afectan otros sistemas neurosensoriales como la audición); o sistémica (con compromiso de otros órganos).
La RP no sindrómica se hereda en forma autosómica dominante (Ad), autosómica recesiva (Ar), o recesiva ligada a X (XL). Se han descrito formas muy infrecuentes de herencia digénica (0,4% de las formas dominantes) y de herencia mitocondrial.
Las características clínicas son muy variables entre pacientes, incluso entre miembros afectados dentro de una misma familia y son en general independientes del gen mutado. A nivel mundial la prevalencia de RP varía entre 1 en 3000 a 1 en 5000 personas por lo que podemos suponer que en Chile habría entre 3400 y 5600 personas con RP. Se estima que la RP da cuenta del 50% de los casos de distrofias retinales hereditarias por lo que el número de pacientes afectados por alguna distrofia retinal hereditaria sería entre 7000 y 11000 pacientes, correspondientes a un 1.4 a 2.2% del total de pacientes con discapacidad visual en el país.

Síntomas
Los síntomas iniciales se relacionan al compromiso de los bastones. Debido a que los bastones se concentran en la medio periferia de la retina y funcionan en condiciones de baja o poca iluminación, su degeneración afecta la visión periférica y la visión nocturna. La ceguera nocturna o nictalopía es uno de los síntomas más precoces de la RP.
Con el paso del tiempo la visión periférica o campo visual se reduce progresivamente. Al avanzar la enfermedad los Conos pueden afectarse, comprometiéndose la agudeza visual, la visión central y la visión de colores.
La RP es diagnosticada típicamente en niños, adolescentes o adultos jóvenes. Es una enfermedad progresiva, aunque la velocidad y grado de progresión y compromiso de la visión varía de persona a persona.
Muchos pacientes afectados de RP son legalmente ciegos alrededor de los 40 años, con un campo visual menor a 20º. - Definición de ceguera legal y discapacidad visual. Link

Modo de Herencia
Se estima que en Chile habrían entre 3 y 5 mil pacientes con RP, la mayoría por mutaciones (variantes patogénicas) en un único gen heredado de uno o ambos padres. El gen mutado entrega instrucciones incorrectas a los fotorreceptores, indicandoles la síntesis de una proteína incorrecta, o pequeña o en algunos casos mucha proteína (las células necesitan una cantidad adecuada de proteínas específicas para poder funcionar adecuadamente). Se han descrito mutaciones en sobre 80 genes relacionados a RP.
Las mutaciones pueden ser transmitidas desde los progenitores a sus hijos a través de 3 diferentes tipos de herencia:
RP autosómica recesiva, en este modo de herencia, ambos padres son portadores de una copia del gen mutado (cada copia del gen es llamado alelo) y una copia normal, y no presentan síntomas de la enfermedad, por lo que son denominados portadores asintomáticos. Cada hijo presenta un 25% de posibilidades de estar heredar una copia mutada de cada progenitor. Si el hijo hereda una copia mutada de uno de los progenitores y una copia sana del otro, será un portador asintomático.
En la forma autosómica dominante de RP, un progenitor es afectado y es el único progenitor con una copia mutada del gen. Cada hijo presenta un 50% de posibilidades de estar afectado al recibir una copia mutada del gen.
En la RP ligada a X, el gen mutado se ubica en el cromosoma X. Las mujeres tienen 2 cromosomas X a diferencia de los hombres que tienen un cromosoma X y un cromosoma Y. Debido a que las mujeres poseen una versión sana del gen en el segundo cromosoma X, las mujeres portadoras son menos afectadas por las enfermedades ligadas a X.
Los hombres al tener solo 1 cromosoma X, son genéticamente susceptibles a enfermedades ligadas al cromosoma X. Los hombres con enfermedades ligadas a X transmiten su cromosoma Y a sus hijos hombres por lo que nunca pasaran una enfermedad ligada a X a sus hijos hombres.
Las portadoras tienen un 50% de riesgo de pasar el cromosoma X con la mutación a sus hijas, quienes serán portadoras y 50% de riesgo de pasar el cromosoma X con la mutación a sus hijos, quienes serán afectados por la enfermedad.
Si un miembro de la familia es diagnosticado con RP, es aconsejable que otros miembros de la familia se realicen un examen por un oftalmólogo entrenado en identificar y tratar enfermedades retinales degenerativas. El asesoramiento genético es importante para discutir el riesgo de la enfermedad en otros miembros de la familia, informar sobre test genéticos y otros temas relacionados.
Vivir con la Enfermedad
Hay abundante información sobre elementos de asistencia para pacientes y familias con RP. Visite el link baja visión.
Diagnóstico Genético
Existe diagnóstico genético para RP. Es útil en confirmar el tipo específico de RP, su modo de herencia cuando no es claro a partir de la historia clínica y por ende el riesgo de transmitirlo a la descendencia. Un paciente con diagnóstico genético está en mejor posición para comprender nuevas estrategias terapéuticas dirigidas a genes y mutaciones específicas y cuáles ensayos clínicos son más apropiados para el.
Ensayos Clínicos
Para saber las novedades sobre recientes avances en RP, puede acceder al siguiente link novedades.
Visite nuestro listado de ensayos clínicos o diríjase directamente a www.ClinicalTrials.gov.
Infografía:
Descargue Infografía “Qué debo saber sobre la Retinitis Pigmentosa”